Scientists Discover 2 Key Brain Systems Behind Psychosis

Researchers have identified dysfunctions in two specific brain systems in people who have psychosis – systems that help us filter attention to important internal and external information and predict or anticipate rewards.
The new study led by a team from Stanford University helps us understand how the symptoms occur, which could lead to better treatments and interventions for psychosis and the other mental health conditions that it’s linked to.
Psychosis involves disconnections from reality, like hallucinations and delusions. It can occur on its own but is also a part of conditions like schizophrenia and bipolar disorder, which scientists are still trying to figure out fully.
“This work provides a good model for understanding the development and progression of schizophrenia, which is a challenging problem,” says cognitive neuroscientist Kaustubh Supekar from Stanford University.
Supekar and his colleagues looked at brain scans of 445 people with conditions including autism, ADHD, early psychosis, and 22q11.2 deletion syndrome (a genetic condition with an increased psychosis and schizophrenia risk). They also looked at scans of 411 healthy people as controls.
Using a machine learning algorithm, the team identified differences in brain function between the groups – leading them to the anterior insula (a crucial part of the salience network filter that helps direct our attention), and the ventral striatum and related dopamine-driven pathways (that control reward-seeking).
While the research group has previously suspected the salience network might be involved, this new study supports that hypothesis and shows that the 22q11.2 deletion syndrome can be a useful way of analyzing psychosis symptoms and risk.
Differences in these areas relating to information filtering and reward prediction were significant in both people with 22q11.2 deletion syndrome and psychosis, and in people with psychosis of unknown origin, the researchers found.
“This parallel reinforces our understanding of psychosis as a condition with identifiable and consistent brain signatures,” says Vinod Menon, a cognitive neuroscientist at Stanford University.
“This process derails the normal functioning of cognitive control, allowing intrusive thoughts to dominate, culminating in symptoms we recognize as psychosis.”
The next step, potentially, is to develop treatments based on these discoveries. Part of the difficulty in treating psychosis is that it can be tricky to see what effects different methods are having on the brain, but now scientists and doctors can be more precise in terms of where they’re looking.
Existing approaches to treating psychosis, including transcranial magnetic stimulation and focused ultrasound, could be adapted to target these particular brain centers in young people at risk of psychosis, the researchers say.
The team is also keen to help reduce the stigma and increase support for those with psychosis. Conditions where people are untethered from reality can be scary to experience, and the line between normal and abnormal neurological function is not as clear as you might think.
“Our discoveries underscore the importance of approaching people with psychosis with compassion,” says Menon.
The research has been published in Molecular Psychiatry.
Source : 1
SCDA urges FSSAI to act on Nestle India for allegedly adding high level sugar in baby foods
 Delhi-based South Chemists & Distributors Association (SCDA), which represents chemists and pharmaceutical distributors working in Delhi, has requested the Food Safety and Standards Authority of India (FSSAI) to take action against multinational corporation Nestle India for allegedly adding high sugar content in baby foods, affecting the health of children. Delhi-based South Chemists & Distributors Association (SCDA), which represents chemists and pharmaceutical distributors working in Delhi, has requested the Food Safety and Standards Authority of India (FSSAI) to take action against multinational corporation Nestle India for allegedly adding high sugar content in baby foods, affecting the health of children.In a letter to ministry of health and family welfare secretary Apurva Chandra and senior officials in the FSSAI, the Association referred to news reports on a study by the International Baby Food Action Network (IBFAN), which alleges that Nestle baby products in poorer countries contain high level of added sugar which are not present in developed countries. Referring to various news and feature reports from various publications, the Association alleged that the infants below 24 months should not be given any added sugar for consumption, but the company is adding sugar which can be to reduce cost of production. It added that increased consumption of added sugar increases risk of type 2 diabetes, obesity, cancer and lifelong chronic illness. Alleging that the company has caused irreparable harm to the children of India the Association pointed that the Sections the Food Safety and Standards Act, 2006 puts the responsibility on FSSAI to monitor manufacturing, processing, distribution, sale and import of food and ensure safe and wholesome food, and limiting the use of food additives. The Act also mentions that FSSAI has the responsibility to achieve an appropriate level of protection of human life and health protection of consumer interests. “As a responsible association, we request action from FSSAI towards Nestle India under Section 54 of the Food Safety and Standards Act 2006 along with any other penal section FSSAI may deem fit. Such action under the existing laws is of utmost requirement as the MNCs take our country for granted and they do not think about our health over their profit motives,” said the letter signed by Yash Aggarwal, legal head of SCDA. The new reports on the study said that Cerelac baby cereals in India have nearly three grams of added sugar per serving and the same products contain five to six grams of sugar per serving in Ethiopia and Thailand respectively. Responding to the allegations in the new reports, Nestle India on April 18 said, “Reduction of added sugars is a priority for Nestlé India. Over the past 5 years, we have already reduced added sugars by up to 30%, depending on the variant. We regularly review our portfolio and continue to innovate and reformulate our products to further reduce the level of added sugars, without compromising on nutrition, quality, safety, and taste.” The company added that it ensures that its products manufactured in India are in full and strict compliance with CODEX standards (a commission established by WHO and FAO) and local specifications (as required) pertaining to the requirements of all nutrients including added sugars. Reports also pointed out that Nestle in its website carries an advice that it is not recommended to add sugar when preparing food for the baby, and not to offer the baby sugary drinks. |
For The First Time, Scientists Showed Structural, Brain-Wide Changes During Menstruation
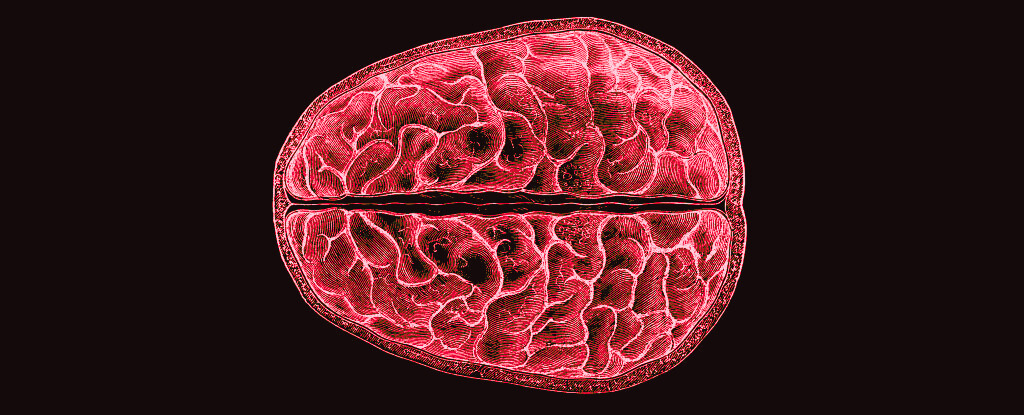
The constant ebb and flow of hormones that guide the menstrual cycle don’t just affect reproductive anatomy. They also reshape the brain, and a study has given us insight into how this happens.
Led by neuroscientists Elizabeth Rizor and Viktoriya Babenko of the University of California Santa Barbara, a team of researchers tracked 30 women who menstruate over their cycles, documenting in detail the structural changes that take place in the brain as hormonal profiles fluctuate.
The results, which are yet to be peer-reviewed but can be found on preprint server bioRxiv, suggest that structural changes in the brain during menstruation may not be limited to those regions associated with the menstrual cycle.
“These results are the first to report simultaneous brain-wide changes in human white matter microstructure and cortical thickness coinciding with menstrual cycle-driven hormone rhythms,” the researchers wrote.
“Strong brain-hormone interaction effects may not be limited to classically known hypothalamic-pituitary-gonadal-axis (HPG-axis) receptor-dense regions.”
People who menstruate will experience some 450 or so periods during the course of their lifetimes, so it would be nice to know the different effects they can have on the body, really.
However, although it is something that happens to half the world’s population for half their lives, research has been somewhat lacking. Who knows why. Total mystery. Seriously.
Most of the research on the hormonal effect on the brain has been focused on brain communication during cognitive tasks, not the actual structures themselves.
“Cyclic fluctuations in HPG-axis hormones exert powerful behavioral, structural, and functional effects through actions on the mammalian central nervous system,” Rizor, Babenko, and their team noted. “Yet, very little is known about how these fluctuations alter the structural nodes and information highways of the human brain.”
The microstructure of white matter – the fatty network of neuronal fibers that transfer information between regions of gray matter – has been found to change with hormonal shifts, including puberty, oral contraception use, gender-affirming hormone therapy, and post-menopausal estrogen therapy.
To address the menstruation gap in our understanding, the team took MRI scans of their subjects during three menstrual phases: menses, ovulation, and mid-luteal. At the time of each of these scans, the researchers also measured the participants’ hormone levels.
The results showed that, as hormones fluctuate, gray and white matter volumes change too, as does the volume of cerebrospinal fluid.
In particular, just before ovulation, when the hormones 17β-estradiol and luteinizing hormone rise, the brains of the participants showed white matter changes suggesting faster information transfer.
Follicle-stimulating hormone, which rises before ovulation, and helps stimulate the ovary follicles, was associated with thicker gray matter.
Progesterone, which rises after ovulation, was associated with increased tissue and decreased cerebrospinal fluid volume.
What this means for the person driving the brain is unknown, but the research lays the groundwork for future studies, and perhaps understanding the causes of unusual but severe period-related mental health problems.
“Although we do not currently report functional consequences or correlates of structural brain changes, our findings may have implications for hormone-driven alterations in behavior and cognition,” the researchers wrote.
“Investigation of brain-hormone relationships across networks is necessary to understand human nervous system functioning on a daily basis, during hormone transition periods, and across the human lifespan.”
The team’s paper is available on bioRxiv.
An earlier version of this article was published in October 2023.
Source : 1
NeuroSmart Portable MER system paves way for improved treatments in Parkinson’s disease
Positive outcomes with brain pacemakers & NeuroSmart Portable MER (micro-electrode recording) system have paved the way for improved treatments in Parkinson’s disease. The advanced technology according to neuro experts represents a significant leap in deep brain stimulation (DBS) therapy with advanced target localization capabilities, automatic navigation, and connectivity tools for offering unparalleled precision in brain analysis.
A medical breakthrough, at the Fortis Hospital, Bengaluru which said that it managed to set a new benchmark by becoming the first medical care centre in India to implement the NeuroSmart Portable MER system for the treatment of Parkinson’s disease.
It is gathered that a 68-year-old patient battling Parkinson’s disease and severe comorbidities, showed recovery, underlining the efficacy and transformative potential of this technology. Discharged just five days post-surgery. Dr Raghuram G, additional director, neurosurgery, Fortis Hospital, said that such a significant improvement underscores a new era in the management and treatment of Parkinson’s disease at Fortis.
NeuroSmart Portable MER system allows doctors see how the patient is responding to the treatment in real time, which makes the surgery more accurate. The interactive approach enhances neurophysiological mapping of the target area, ensuring precise localization for deep brain stimulation electrode placement. This sophisticated analysis allows us to discern the precise area for electrode placement, ensuring optimal therapeutic efficacy and minimizing potential side effects for our patients, he added.
According to Dr Guruprasad Hosurkar, additional director neurology, Fortis Hospital, said that DBS had a profound effect on enhancing the quality of life for a patient. By alleviating tremors and reducing stiffness, DBS enabled the patient to lead an independent life, empowering him to regain control and enjoy improved well-being.
Noting that global burden of Parkinson’s disease exceeds 10 million, of which India alone accounts for a substantial 10%, Rajinish Menon, CEO & founder, Sukino Healthcare Solutions said that the increasing incidence of the condition in India, especially among the youth population, is a crucial reason why we needs to step up efforts to develop accessible and effective treatments for Parkinson’s.
Currently, Parkinson’s is incurable. Early detection, along with continuous rehabilitation and holistic, personalised treatment, remains the most practical choice. Although research has uncovered that implanting brain pacemakers presents a long-term management option for Parkinson’s, this avenue is inaccessible due to the high costs involved, he added.
Source : 1
A more affordable, easily available treatment approach is continuum care, with services ranging from medication management to emotional support. This offers lasting effects, such as improvements in physical health and the promotion of neuroplasticity, which are crucial for slowing Parkinson’s-related brain degeneration, said Menon.
Doctor Reveals What Happens When You Stop Taking Ozempic
Drugs like Ozempic are very effective at helping most people who take them lose weight. Semaglutide (sold as Wegovy and Ozempic) and tirzepatide (sold as Zepbound and Mounjaro) are the most well known in the class of drugs that mimic hormones to reduce feelings of hunger.
But does weight come back when you stop using it?
The short answer is yes. Stopping tirzepatide and semaglutide will result in weight regain in most people.
So are these medications simply another (expensive) form of yo-yo dieting? Let’s look at what the evidence shows so far.
It’s a long-term treatment, not a short course
If you have a bacterial infection, antibiotics will help your body fight off the germs causing your illness. You take the full course of medication, and the infection is gone.
For obesity, taking tirzepatide or semaglutide can help your body get rid of fat. However it doesn’t fix the reasons you gained weight in the first place because obesity is a chronic, complex condition. When you stop the medications, the weight returns.
Perhaps a more useful comparison is with high blood pressure, also known as hypertension. Treatment for hypertension is lifelong. It’s the same with obesity. Medications work, but only while you are taking them. (Though obesity is more complicated than hypertension, as many different factors both cause and perpetuate it.)
Therefore, several concurrent approaches are needed; taking medication can be an important part of effective management but on its own, it’s often insufficient. And in an unwanted knock-on effect, stopping medication can undermine other strategies to lose weight, like eating less.
Why do people stop?
Research trials show anywhere from 6% to 13.5% of participants stop taking these drugs, primarily because of side effects.
But these studies don’t account for those forced to stop because of cost or widespread supply issues. We don’t know how many people have needed to stop this medication over the past few years for these reasons.
Understanding what stopping does to the body is therefore important.
So what happens when you stop?
When you stop using tirzepatide or semaglutide, it takes several days (or even a couple of weeks) to move out of your system. As it does, a number of things happen:
- you start feeling hungry again, because both your brain and your gut no longer have the medication working to make you feel full
- blood sugars increase, because the medication is no longer acting on the pancreas to help control this. If you have diabetes as well as obesity you may need to take other medications to keep these in an acceptable range. Whether you have diabetes or not, you may need to eat foods with a low glycemic index to stabilise your blood sugars
- over the longer term, most people experience a return to their previous blood pressure and cholesterol levels, as the weight comes back
- weight regain will mostly be in the form of fat, because it will be gained faster than skeletal muscle.
While you were on the medication, you will have lost proportionally less skeletal muscle than fat, muscle loss is inevitable when you lose weight, no matter whether you use medications or not. The problem is, when you stop the medication, your body preferentially puts on fat.
Is stopping and starting the medications a problem?
People whose weight fluctuates with tirzepatide or semaglutide may experience some of the downsides of yo-yo dieting.
When you keep going on and off diets, it’s like a rollercoaster ride for your body. Each time you regain weight, your body has to deal with spikes in blood pressure, heart rate, and how your body handles sugars and fats. This can stress your heart and overall cardiovascular system, as it has to respond to greater fluctuations than usual.
Interestingly, the risk to the body from weight fluctuations is greater for people who are not obese. This should be a caution to those who are not obese but still using tirzepatide or semaglutide to try to lose unwanted weight.
How can you avoid gaining weight when you stop?
Fear of regaining weight when stopping these medications is valid, and needs to be addressed directly. As obesity has many causes and perpetuating factors, many evidence-based approaches are needed to reduce weight regain. This might include:
- getting quality sleep
- exercising in a way that builds and maintains muscle. While on the medication, you will likely have lost muscle as well as fat, although this is not inevitable, especially if you exercise regularly while taking it
- addressing emotional and cultural aspects of life that contribute to over-eating and/or eating unhealthy foods, and how you view your body. Stigma and shame around body shape and size is not cured by taking this medication. Even if you have a healthy relationship with food, we live in a culture that is fat-phobic and discriminates against people in larger bodies
- eating in a healthy way, hopefully continuing with habits that were formed while on the medication. Eating meals that have high nutrition and fibre, for example, and lower overall portion sizes.
Many people will stop taking tirzepatide or semaglutide at some point, given it is expensive and in short supply. When you do, it is important to understand what will happen and what you can do to help avoid the consequences. Regular reviews with your GP are also important.
Natasha Yates, General Practitioner, PhD Candidate, Bond University
This article is republished from The Conversation under a Creative Commons license. Read the original article.
Source : 1
